
Werner syndrome/RecQ helicases
 Background: The human RecQ helicases are encoded by
a small, 5-member gene family. All five RecQ proteins contain a conserved
RecQ helicase concensus domain, together with one or more additional conserved
domains. Heritable mutations in the human WRN, BLM and RTS/RECQL4
genes lead to the genetic instability/ cancer predisposition syndromes Werner syndrome,
Bloom syndrome and Rothmund-Thomson syndrome respectively. Werner syndrome is the only
RecQ helicase deficiency syndrome whose phenotype mimics premature aging. WRN is the
only human RecQ helicase protein that has exonuclease as well as helicase activity.
Background: The human RecQ helicases are encoded by
a small, 5-member gene family. All five RecQ proteins contain a conserved
RecQ helicase concensus domain, together with one or more additional conserved
domains. Heritable mutations in the human WRN, BLM and RTS/RECQL4
genes lead to the genetic instability/ cancer predisposition syndromes Werner syndrome,
Bloom syndrome and Rothmund-Thomson syndrome respectively. Werner syndrome is the only
RecQ helicase deficiency syndrome whose phenotype mimics premature aging. WRN is the
only human RecQ helicase protein that has exonuclease as well as helicase activity.
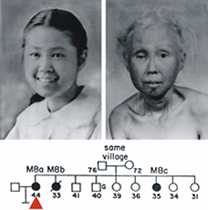
The two photos shown below left are of a Japanese-American Werner syndrome patient as a teenager, and 30 years later as a woman in her mid-40's. This patient and her sister (see pedigree below the photos) were seen at the University of Washington in Seattle in the early 1960's. This now-iconic clinical photo pair shows the striking age- and time-dependent changes seen in Werner syndrome patients. These changes begin in the 2nd decade of life, and are accompanied by the development of clinically important, age-associated diseases such as cancer, atherosclerotic cardiovascular disease, diabetes mellitus and osteoporosis. Patients may die prematurely of either cancer or accelerated cardiovascular disease. A full description of the Werner syndrome phenotype can be found in recent publications from the lab, at the International Registry of Werner Syndrome website, or in the Online Mendelian Inheritance in Man (OMIM) listing for Werner syndrome at the National Library of Medicine.

 Protein function: The Werner syndrome protein has 3'-to-5' helicase
and exonuclease activities. The physiologic role(s) of WRN are not known in any detail, although loss of
WRN function can promote aberrant homology-dependent recombination, mutagenesis and telomere replication defects.
Protein function: The Werner syndrome protein has 3'-to-5' helicase
and exonuclease activities. The physiologic role(s) of WRN are not known in any detail, although loss of
WRN function can promote aberrant homology-dependent recombination, mutagenesis and telomere replication defects.
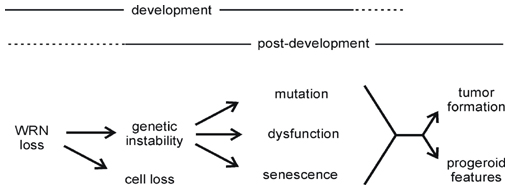
The aim of ongoing research is to delineate the molecular basis of WRN function in human and mouse cells, and to analyze functional crosstalk or redundancy among members of the human RecQ helicase family. We are also interested in determining how heritable or acquired changes in WRN expression may alter disease risk or the response to therapy. Our long term goal is to develop a sophisticated molecular model of WRN function at the cellular level, and then use this understanding to explain how a loss of WRN function contributes to cell lineage-specific disease pathogenesis. A simple model of WRN disease pathogenesis is shown below. For more detailed discussion see our recent reviews on Werner syndrome.
WRN Mutation database: We maintain the Werner Syndrome Locus-Specific Mutational Database for the Human Genome Organization and the Human Genome Variation Society. This database provides detailed information on WRN mutations identified in clinically ascertained Werner syndrome patients. The database was updated in Summer 2007 to include the large number of new mutations reported in 2006 and 2007. The symbols in the figure below summarize the locations of mutations identified in Werner syndrome patients in the WRN protein and mRNA. The open and filled symbols in the figure below identify two large sets of mutations originally characterized by different investigators (see Mutation Database site for more detail).
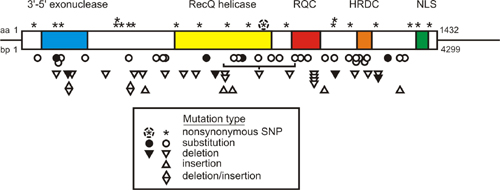
A key feature of all Werner syndrome-associated WRN mutations is that they truncate and promote the loss of WRN protein. This finding, together with functional phenotyping of WRN missense mutants that selectively lack exonuclease or helicase activity, indicate that the cellular phenotype of Werner syndrome develops only when both WRN catalytic activities are lost from cells. This loss also causes the clinical phenotype.
Our work on Werner syndrome is supported by a Program grant from the NCI on 'Genetic Instability in Werner Syndrome'.
Homing endonucleases
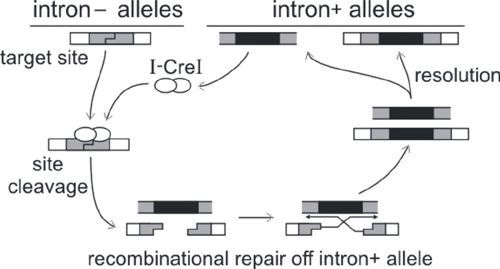
Background: Homing endonucleases are highly site-specific DNA double strand endonucleases that catalyze and target the lateral transfer of parasitic DNA elements. These elements are often self-splicing introns or inteins that encode the homing endonuclease open reading frame. The lateral transfer or 'homing' cycle for mobile introns and inteins is shown below in outline. The net result is the unilateral, and irreversible, gain of endonuclease-encoding introns by previously intron-minus alleles.
There are four major families of homing endonuclease proteins. We have focused on structural and functional analysis of members of the large LAGLIDADG family and the smaller His-Cys box family. The family names are derived from conserved sequence motifs found in each family and family member.
Structural biology: We began, over a decade ago with collaborator Barry Stoddard, to obtain high resolution co-crystal structures of homing endonuclease proteins bound to their target or homing site DNA's. Barry and his lab have subsequently determined high resolution co-crystal structures of members of all of the homing endonuclease families [see Stoddard Lab for additional information].
The high resolution co-crystal structure of the LAGLIDADG homing endonuclease I-CreI is shown below, together with a higher power view of the dimer/domain folding interface alpha helices and three-metal active site characteristic of members of this protein family.
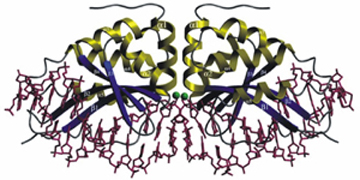
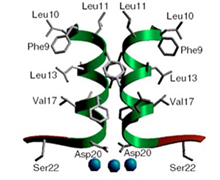
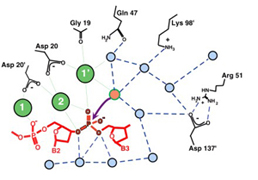
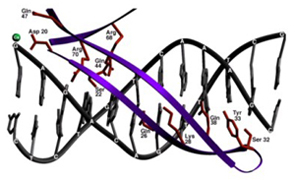
Engineering: Homing endonucleases have the remarkable property of binding and cleaving single genes in complex genomes without the aid of additional proteins or cofactors apart from magnesium. High resolution structures of several homing endonucleases have revealed how this feat of molecular recognition is achieved by a small protein. These structural data also guide our current efforts to modify the DNA target site specificity of homing endonucleases, using experimental and computational approaches.
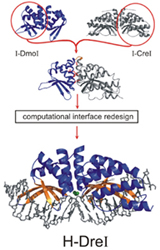
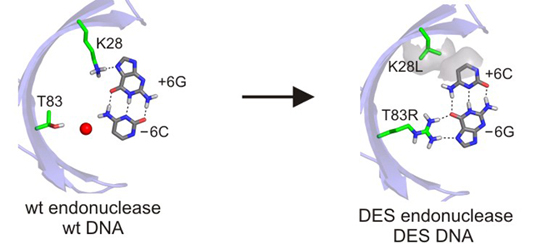
Two examples of LAGLIDADG homing endonuclease engineering, performed with the Stoddard and Baker labs, are shown below. We've engineered a novel chimeric LAGLIDADG homing endonuclease (H-DreI, shown below at left) from a subunit of I-DmoI and a monomer of I-CreI (Chevalier et al. (2002) Molecular Cell 10:895-905). We have also shown that computational design can be used to alter the DNA recognition specificity of I-MsoI, another LAGLIDADG homing endonuclease (Ashworth et al. (2006) Nature 441:656-659). Thus far we have shown that novel chimeric LAGLIDADG homing endonuclease proteins can be engineered (H-DreI), and that computational design can be used to alter the DNA recognition specificity of I-MsoI, a LAGLIDADG homing endonuclease (shown below at right).
Gene targeting: We are now developing gene-specific variants of homing endonucleases to facilitate targeted gene correction or gene ablation in human and insect cells. The aim of work with colleagues in the newly funded Northwest Genome Engineering Consortium (NGEC) is high efficiency, targeted recombinational repair of disease-causing mutations in hematopoietic or lymphoid cells. The diseases we are focusing on are X-linked severe combined immunodeficiency syndrome (X-SCID), Bruton agammaglobulinemia (BTK or XLA), red cell defects associated with mutations in the ß-globin (HBB) or pyruvate kinase (PKLR) genes, and the most common form of tyrosinemia (FAH mutations).
This work is supported by a new U54 Interdisciplinary Research Consortium grant from the NIH, coordinated by the Northwest Genome Engineering Consortium (NGEC).
 Disease prevention: We are also developing novel homing endonuclease variants to prevent
malaria. This work is being done with colleagues in Seattle, at Imperial College London, Cambridge University,
Oxford University and at the London School of Hygiene and Tropical Medicine. Anopheles mosquito gene-specific
homing endonucleases are being developed to cleave and inactivate mosquito genes required for malaria parasite
development or for the generation of fertile biting females. These two strategies in concert should disrupt the
transmission of malaria and provide a new and effective way to control the Anopheles mosquitos that propagate malaria.
Disease prevention: We are also developing novel homing endonuclease variants to prevent
malaria. This work is being done with colleagues in Seattle, at Imperial College London, Cambridge University,
Oxford University and at the London School of Hygiene and Tropical Medicine. Anopheles mosquito gene-specific
homing endonucleases are being developed to cleave and inactivate mosquito genes required for malaria parasite
development or for the generation of fertile biting females. These two strategies in concert should disrupt the
transmission of malaria and provide a new and effective way to control the Anopheles mosquitos that propagate malaria.
Our work on malaria prevention is being done with funding from a Gates Grand Challenges in Global Health grant administered by the Foundation for the NIH.
Analysis of DNA replication
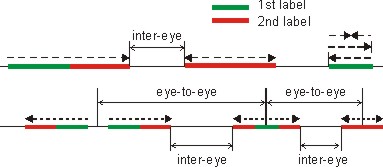
New protocols: A wide variety of assays have been developed to analyze DNA replication by examining genomic DNA, chromosomes or individual genes. These complementary assays have been used to gain mechanistic insight and to identify genetic, environmental and pharmacologic perturbations that alter DNA replication. In our lab, we focus on technologies that allow visualization of individual replicating DNA molecules. At the core of these technologies lies the ability to pulse-label replicating genomic DNA in vivo with halogenated pyrimidine nucleoside analogs such as bromo-deoxyuridine, BrdU, and then stretch isolated DNA molecules to approximately half the contour length (1 µm =4Kb) and affix them to glass support for staining and fluorescent microscopy. As a result, analogs incorporated into DNA by replisomes show as linear tracts within a DNA fiber, and their lengths and distribution can be easily measured and quantified. Sequential labeling with pairs of analogs (e.g. chloro- and iodo-deoxyuridine) followed by detection with analog-specific antisera gives the method additional versatility and provides a powerful way to reveal replication dynamics in vivo. For example, this approach allows comparing progression of individual replication forks on DNA under normal conditions and under genotoxic stress.
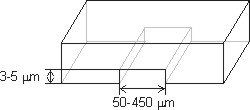
Microfluid capillary channels: Several single molecule replication protocols have been developed. All of them share common labeling and detection protocols, though differ in their solution to the problematic step of reproducibly stretching DNA. We recently developed a facile new solution to this problem by using microfluidic capillary channels to stretch replicating DNA molecules. This protocol, termed microfluidic-assisted replication track analysis, ma-RTA, builds on earlier work by the lab of David Schwartz, in which a similar approach was used for restriction site mapping of prokaryotic and eukaryotic genomes. Our microfluidic platform consists of parallel arrays of 3-sided, 3 or 4 µm high, 150-450 µm wide, and 1cm long capillary channels fabricated from polymethyl siloxane (PDMS) by conventional soft lithography, and silane-modified glass coverslips to reversibly seal the open side of the channels. Capillary tension in these microchannels facilitates DNA loading, stretching and deposition onto coverslip glass from µL-scale DNA samples. A detailed discussion of the protocol and a step-by-step guide are available online as a 2009 Nature Protocols publication.

WS-WRN: We recently used ma-RTA to determine the role of the RecQ helicase WRN, mutated in the human Werner Syndrome, in replication during genotoxic stress (link). This study showed that WRN depletion in human fibroblasts significantly reduced the speed at which replication forks elongated in vivo after alkylation damage to DNA by MMS or replication arrest by HU treatment. These results establish the importance of WRN during genomic replication and indicate that WRN acts to facilitate fork progression under conditions of replication stress. For more detailed discussion of this and related topics please see our publications,in particular the 2008 Cell Cycle paper and 2008 DNA repair review.
Human somatic mutation
HPRT mutagenesis work: Our first work on Werner syndrome was a quantitative and molecular analysis of spontaneous mutations in the X-linked HPRT (hypoxanthine guanine phosphoribosyltransferase) gene in Werner syndrome fibroblast cell lines (Fukuchi et al. (1989) PNAS 86:5893-5897). Although we no longer work actively on HPRT mutagenesis, we have included our most useful protocols and data to aid others working in this important area (see Links for protocols and data).
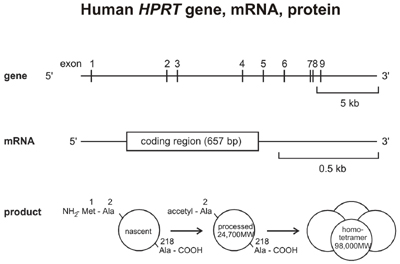
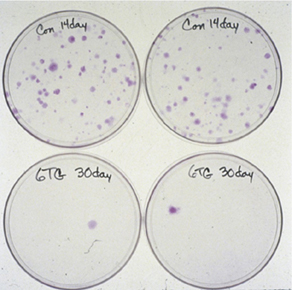
Several years ago the methods developed to isolate and analyze HPRT mutagenesis in human fibroblast and tumor cell lines were extended to an analysis of spontaneous HPRT mutagenesis in primary tubular epithelial cells from human kidney. These analyses showed that spontaneous HPRT mutations were remarkably common in human kidney epithelial cells and could be recovered by simple somatic cell genetic techniques (plate photos below at left), and included a high proportion of new mutations and multiple mutations that had not been previously documented in human somatic cells. The frequency of HPRT mutants in kidney as a function of donor age is shown below right, together with comparison data from human peripheral blood T-lymphocytes.
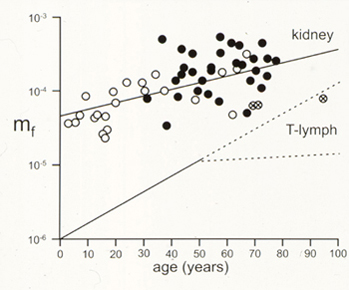
Our HPRT mutation analysis in human kidney has been combined and analyzed with two additional large human HPRT mutation data sets: peripheral blood T-lymphocyte HPRT mutations and germ line HPRT mutations from Lesch-Nyhan syndrome or gout patients. These mutation data sets are available as a spreadsheet for further analysis.
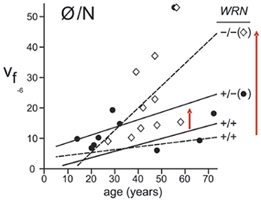
Red blood cell somatic mutation: We have used the red blood cell glycophorin-A mutation assay to quantify somatic mutations arising during red blood cell development in Werner syndrome patients and mutation-typed pedigree members. The assay uses monoclonal antibodies to detect and distinguish between the two major GPA alleles found on human red cells, the M and N blood group alleles. The assay is shown in outline below at left, together with common causes of allelic variants.
Our results document a strong, age-dependent increase in GPA allele loss (N/Ř) variants in Werner syndrome patients, and elevated N/Ř allele loss variants in WRN mutation-typed heterozygous carriers (see mutant frequency data as a function of genotype and age below at right). WRN mutation carriers are common worldwide, and may be at elevated risk for disease or adverse responses to therapy.

Our work on human somatic mutation was funded by grants from the NCI and NIA.